| 1. | Place the line cursor in a sequence above which the Annotation line is supposed to occur. | 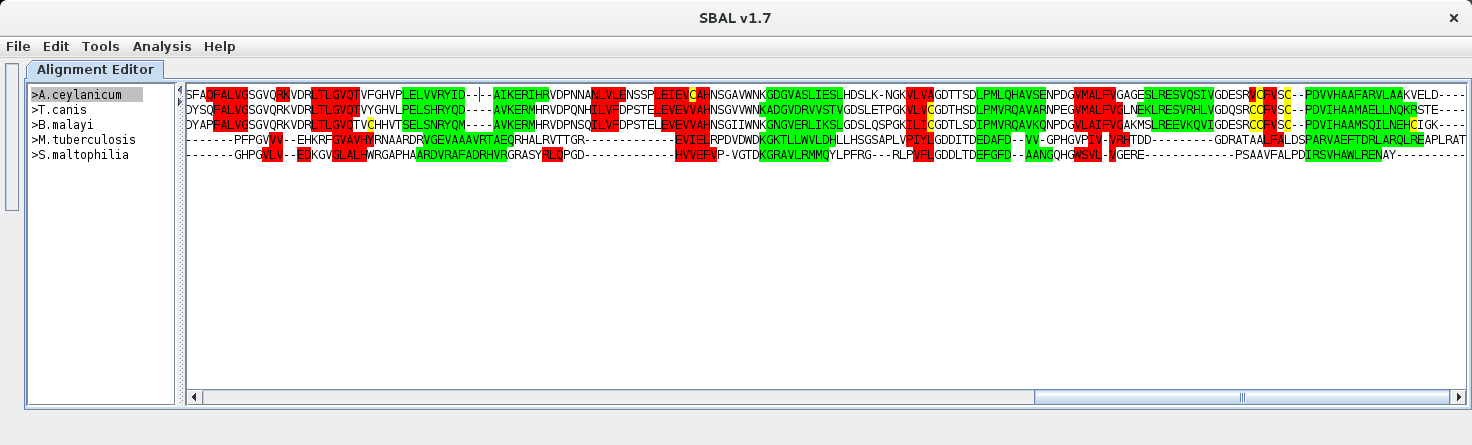 |
| 2. | Use the menu item Edit - Add annotation to generate the annotation line. | 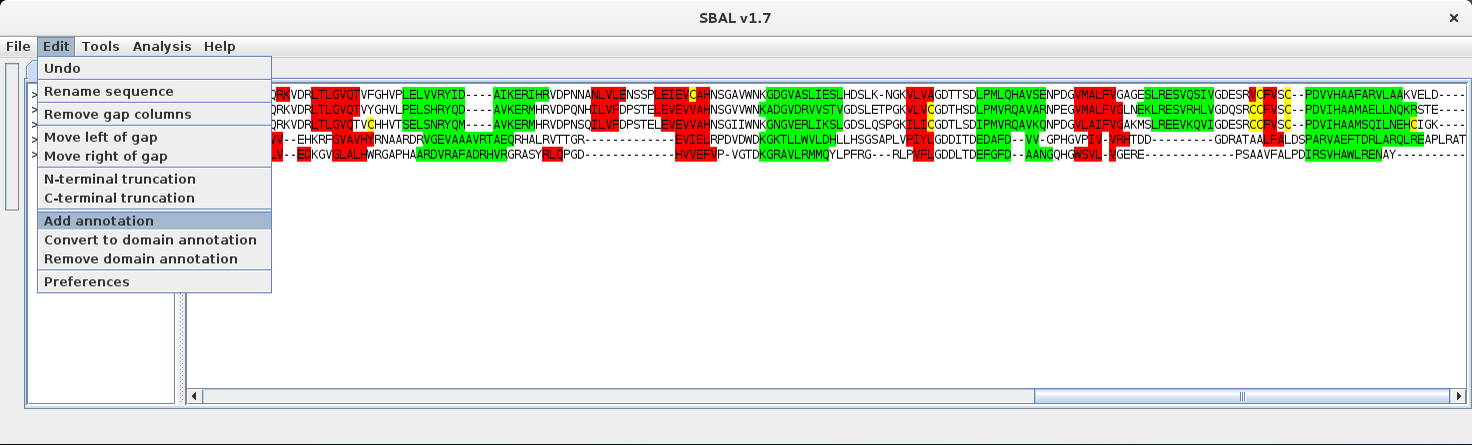 |
| 3. | Enter the annotation text in the newly generated line and use the pipe character | to denote domain boundaries. Place the line cursor in the annotation line. |
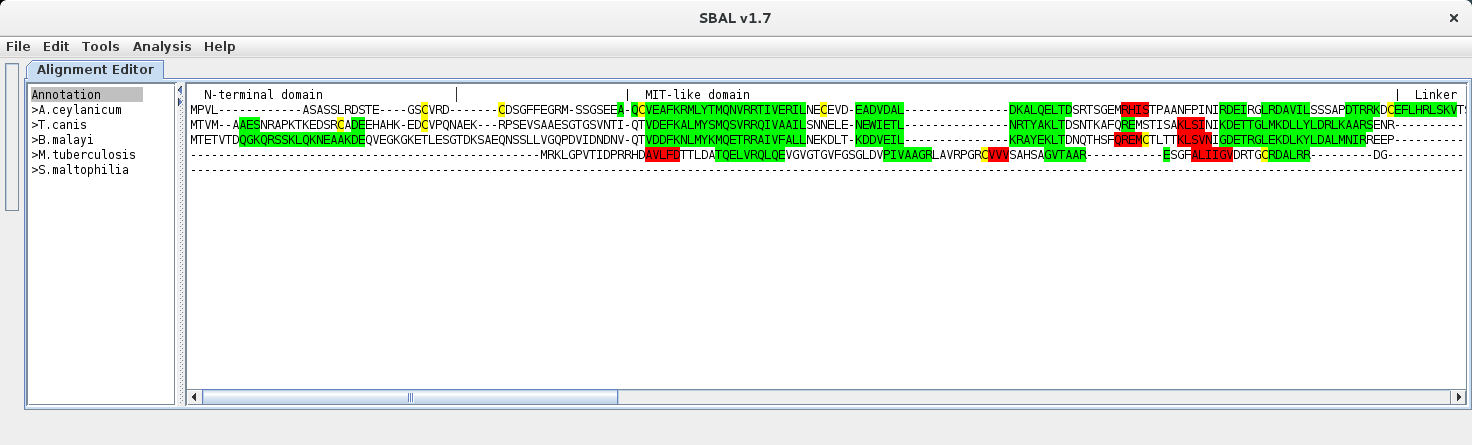 |
| 4. | Then use the menu item Edit - Convert to domain annotation. | 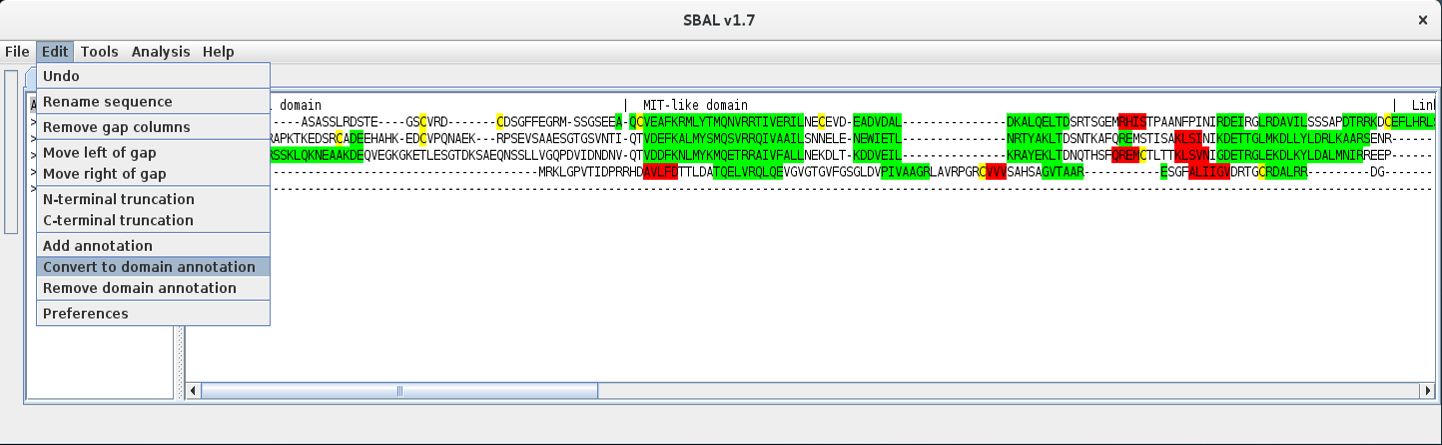 |
| 5. | A popup window with the recognised domains appears. | 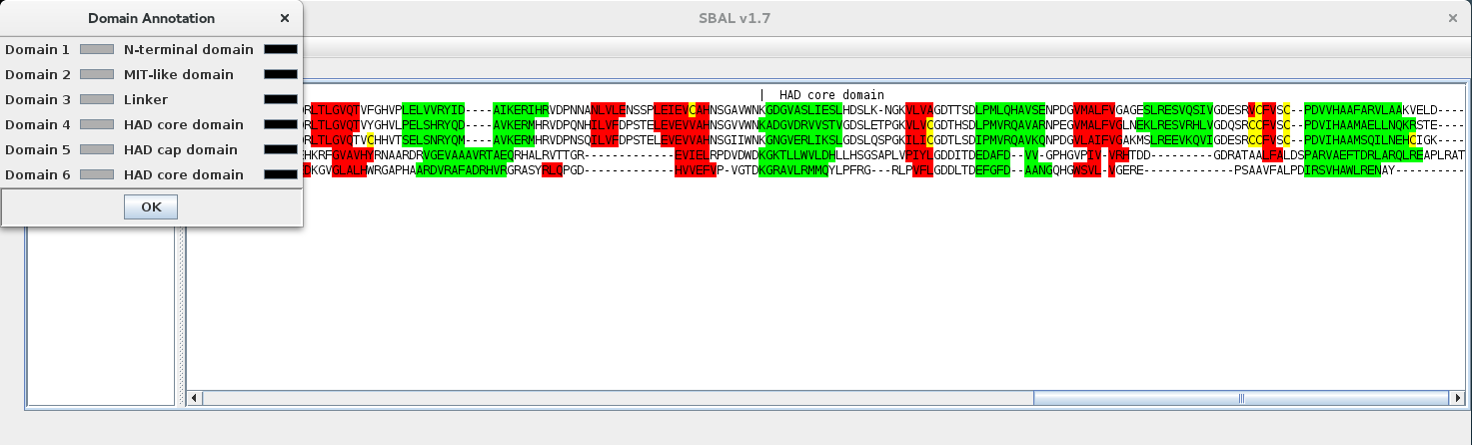 |
| 6. | In the popup window, the background colour (first colour box) of the individual domains, as well as the text colour (second colour box) of the domain description can be selected by clicking on the colour boxes. The annotation text cannot be changed in the popup window. |  |
| 7. | After confirming the choices, the individual domains are shown in the chosen colours. To change any settings, simply repeat from step 1. | 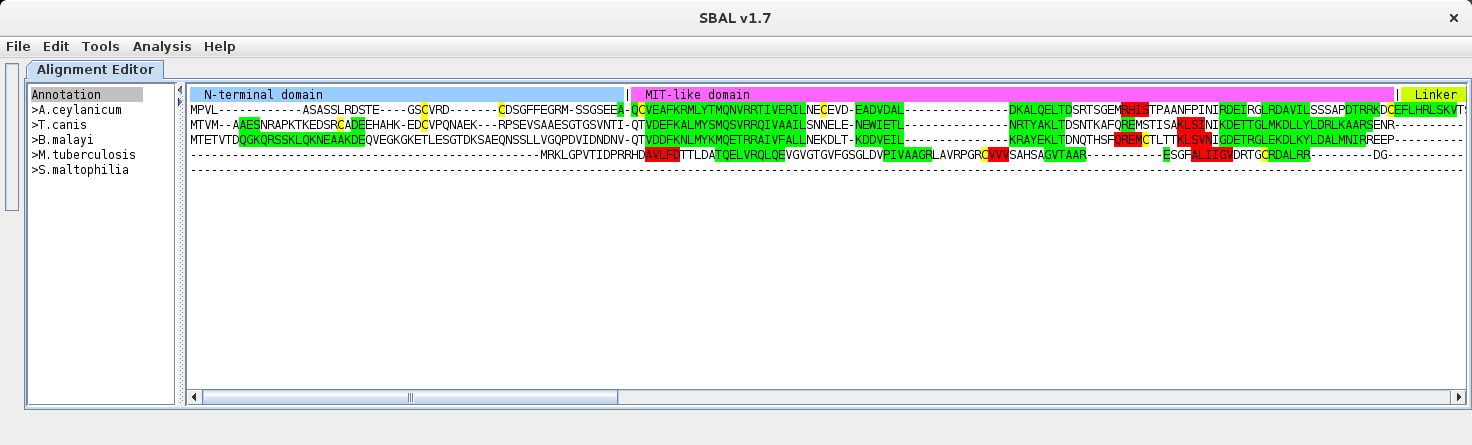 |